ha
Gravimetric Analysis
impurity in crystal lattice where ions are attached to different amount of other ions. For example ag+ attaches to 5 Cl- on the surface and 4 on the edge but 6 in the middle giving a slight partial positive charge.
Cause impurity (coprecipitates)
inclusion: ion with the same charge and size similar to lattice ion is substituting into lattice structure, will make precipitate’s mass higher than expected
difficult to remove because part of lattice structure
reprecipitation removes it which heats and cools to keep reforming precipitate
occlusion: interfering ions become trapped within the growing precipitate, localized in one place
increases mass but can be smaller
minimize by maintaining equilibrium with precipitate in supernatant solution called digestion
adsorption: ions are surface adsorbates and are absorbed
minimize by decreasing precipitates Surface Area
Interferrent: produces a second precipitate
minimize by controlling solution conditions
Mass Charge Balance
Solutions
http://chemed.chem.purdue.edu/genchem/topicreview/bp/ch7/lattice.html
The lattice energy of a salt therefore gives a rough indication of the solubility of the salt in water because it reflects the energy needed to separate the positive and negative ions in a salt.
Sodium and potassium salts are soluble in water because they have relatively small lattice energies. Magnesium and aluminum salts are often much less soluble because it takes more energy to separate the positive and negative ions in these salts. NaOH, for example, is very soluble in water (420 g/L), but Mg(OH)2 dissolves in water only to the extent of 0.009 g/L, and Al(OH)3 is essentially insoluble in water.
The force of attraction between oppositely charged particles is directly proportional to the product of the charges on the two objects (q1 and q2) and inversely proportional to the square of the distance between the objects (r2).
The strength of the bond between the ions of opposite charge in an ionic compound therefore depends on the charges on the ions and the distance between the centers of the ions when they pack to form a crystal.
An estimate of the strength of the bonds in an ionic co
PHYSICAL PROPERTIES OF SOLUTIONS
Concentrations Solution
Molarity (mol solute/ L soln)
Molality (g solute/ kg solvent)
Mol fraction (mol component/ mol solution)
Ppm = mg/L
Ppb = micro(g)/L
Ppt = nano(g)/L
Enthalpy and Intermolecular forces in Solutions
Enthalpy
Enthalpy increases when you hypothetically separate pure components of solute and solvent molecules
Enthalpy decreases as energy is released when solute and solvent combine to make solution
When solution forms, it is endothermic with weaker solute solvent forces when ΔH of solution is greater than 0, exo if less than 0 (stronger solute-solvent forces). Ideal solution is equal to 0.
ΔHsoln = ΔH1 + ΔH2 +ΔH3
Intermolecular Forces
intermolecular forces between solvent molecules determine enthalpy change ΔH1 which is separating solvent molecules
Intermolecular forces between solute is ΔH2 where separating solute molecules
Intermolecular forces between solute and solvent is ΔH3 where solution forms from two separate components.
Comparing these intermolecular forces
If all three forces are same type and comparable in strength, ΔHsoln = 0, volume is the sum of both components. This is Ideal solution (mainly ideal gas mixtures)
If solute-solvent force is stronger, ΔH3 magnitude exceeds sum of other two enthalpy and heat is released (exo). This is why when mixed, solution volume is less than the sum of both component volumes. Nonideal
solute-solvent weaker: ΔH3 magnitde is less than sum of other enthalpy. Nonideal and endo
If solute-solvent is too weak: ΔH3 magnitude is too small making the solute not dissolve in solvent. Heterogenous mixture.
polar and nonpolar don’t form because, for ex. gas has nonpolar hydrocarbon with dispersion, water with hydrogen bonds. no hydrogen bond forms with water and hydrocarbon
Aqueous ionic solutions
attraction of water dipoles with ions pulls ions out of crystal lattice and into aqueous.
ions are hydrated with water molecules that surround
the amount an ionic solid can dissolve depends on competition between the ion-dipole force and interionic attractions holding the ions in a crystal lattice.
if ion-dipole is greater, it is soluble
Equilibrium Solution Formation
liquid mix in all proportions is miscible (no limit) like ethanol and water
dynamic equilibrium in solution is rate of crystallization equals rate of dissolving.
saturated: dynamic equilibrium between undissolved solute and a solution
solubility changes with temperature (very few such as some sulfates decrease with increase temp)
(when cooling saturated solution) supersaturated contains more solute than it would at equilibrium. Unstable, addition of a seed crystal of solute causes all the excess solute to crystallize
crystallizing a solute from concentrated solution by lowering temp can purify substance
Gas Solubilities
most gas become less soluble in liquid as temp increases
at constant temp, the solubility is directly proportional to the pressure of the gas.
S=k*Pgas
Henry’s Law
Gas molecules collide with surface of liquid, as pressure increases more number of molecules per volume in gas, which makes more collisions occur, more gas molecules dissolve.
Colligative Properties (FOR NONELECTROLYTES): depend on the number of solute particles present but not on the identity of the solute
Book:
Vapor Pressures of Solutions
Raoult’s law (For NON-volatile solutes)
presence of solute lowers the vapor pressure of solvent in a solution.
The vapor pressure of the solvent above a solution (Psolv) is the product of the vapor pressure of the pure solvent (Posolv) and the mole fraction of the solvent in the solution (xsolv)
Psolv=xsolv* Posolv
Presence of solute means xsolv<1
Law only usually works for ideal solutions or dilute nonideals
Any Volatile component in a solution
Raoult’s law works as long its ideal solution.
total pressure is sum of two partial pressures.
Psolution = P°AχA + P°BχB
Two volatile component solution
the vapor in equilibrium with an ideal solution of two volatile components has a higher mole fraction of the more volatile component than is found in the liquid.
the new vapor after condendsing a vapor to liquid and vaporizing it would have a higher mole faaction of the more volatile substances (higher vapor pressure). If this is done multiple times a pure substance of that high volatile substance would be obtained.
Fractional distillation
constant pressure and boil the solution in an apparatus where the more volatile substance would be condensed from the vaprot at the top of the column and the less would be retained in the warmer flask.
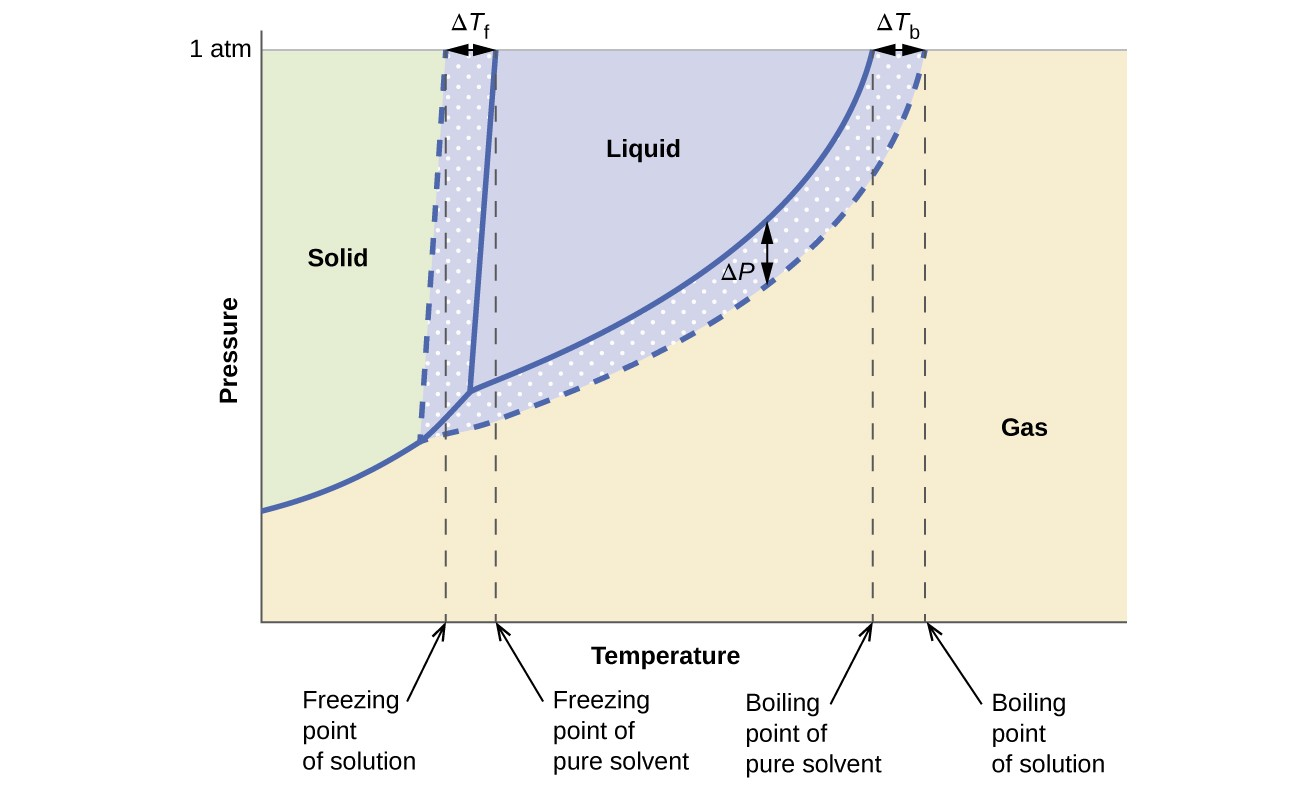
Freezing Point Depression and Boiling Point Elevation
The amount of change is related to the mole fraction of solute present
In dilute solutions, molality is proportional to mole fractions
m represents molality or solute
Kf (cryoscopic) and Kb (ebullioscopic) are when the solute concentration is 1 m
molal (freezing point depression)/(boiling point elevation) constant
The presence of a solute lowers/depresses the freezing point by the quantity of ΔTf
ΔTf = Tf(solution) – Tf(solvent) = -Kf * m
The presence of a solute raises/elevates the boiling point by the amount ΔTb
ΔTb = Tb(solution) – Tb(solvent) = Kb * m
Because solutes remain in solutions and only solvent freeze or boil, the concentration of solute increases as freeze and boiling occur. Therefore freeze and boiling points are not constant and are when the first bit of solution starts to freeze or boil
Osmotic Pressure
Permeable: allow liquids or gases to pass
impermeable: keeps from passing
semipermeable: allow solvent molecules to pass but not solute molecules
Membranes usually sheets or films containing a network of microscopic holes/pores through which small solvent molecules can pass, but larger solute cannot
A natural tendency for solvent molecules to migrate from a region where they exit in greater numbers (low solute mole fraction) to a region where the solvent molecules are less numerous (higher solute mole fraction)
Osmosis: net flow of solvent molecules through a semipermeable membrane from low to high concentration of solution.
Reduce osmosis by applying pressure to force solvent out of solution
Osmotic Pressure: pressure to stop osmosis of solution
Calculating osmotic pressure of dilute solutions of nonelectrolytes is = nVRTwhere pi is osmotic pressure in atm, n is number of moles of solute, V is liters of soln, R is universal gas constant, T is kelvin temp. molarity (M) =nVso it is =MRT
NOTE: Osmotic pressure measurements work well when freezing point depression doesn’t.
Isotonic solution: a solution having the same osmotic pressure as body fluids
Hypertonic solution: a greater concentration of solute causing water to flow out of cells making them shrink. Higher osmotic pressure than cells.
Hypotonic solution: lower osmotic pressure. lower concentration of solute in solution surrounding cells.
reverse osmosis is when the pressure is greater than the amount needed to stop osmosis
Online:
Freezing Point Depression and Boiling Point Elevation
change in temperature is proportional to molality
Vapor Pressure Lowering
Non Volatile
vapor pressure is proportional to the mole fraction, Raoult’s Law
For Pure solvent where mole fraction is 1, the pressure doesn’t change
When there are multiple solutes and mole fraction is less than 1, the vapor pressure of the solvent becomes less than the vapor pressure above the solvent.
Volatile
vapor pressure above solution is equivalent to sum of vapor pressures of solvent and each solute.
Solutions of Electrolytes (Colligative properties)
Colligative properties must base the solute concentration on the total number of particles in soln (ions and/or molecules)
van’t Hoff factor (i) modifies the equations for colligative properties by accounting for presence of ions
Freezing point depression
ΔTf = –i * Kf * m
Boiling point elevation
ΔTb = i * Kb * m
Osmotic pressure
=iMRT
Nonelectrolyte solutions
i = 1, molarity and molality of solute is of the solute particles
Strong electrolyte
i = moles of ions for each mole of solute
ex. NaCl: i = 2, CaCl2: i = 3
As concentration gets more dilute, i approaches integer, else it less than the integer for greater concentrations
singly charged ions solute (like NaCl) are more approach quickly to integer because interionic attractions
attractive forces between cations and anions cause some to associate into ion pairs
Weak electrolyte
1 < i < 2
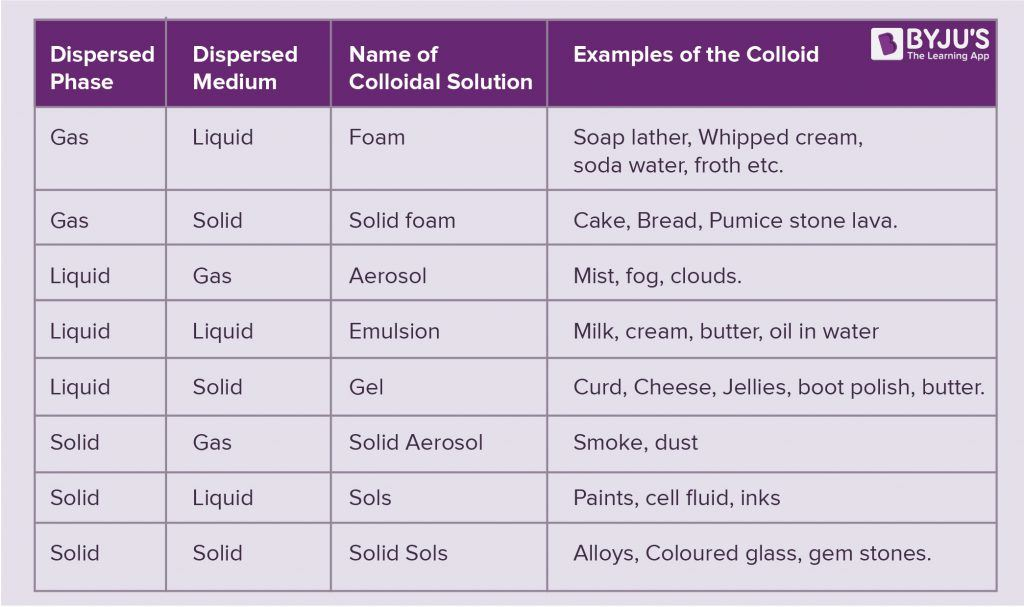
Colloids
homogeneous mixture is a solution (solute and solvent particles less than 1nm diameter)
heterogeneous solution that doesn’t mix is suspension (particle 1000nm or more)
colloid is a dispersion in an appropriate medium of particles ranging from 1nm to 1000nm in size
sols and emulsions are most common
John Tyndall made Tyndall effect which is colloidal nature is shown by scattering a beam of light through the colloid.
Special property of surfaces is to adsorb/attach ions from solution
A high concentration of an electrolyte can cause a colloid to coagulate/precipitate by neutralizing the charges on the colloidal particles
Excess ions can be removed through dialysis (semipermeable membrane) prevent coagulation making colloid more stable
electrodialysis has the process by the attractions of ions to an electrode having the opposite charge
“An azeotrope is a mixture of two or more liquids (chemicals) in such a ratio that its composition cannot be changed by simple distillation. This occurs because, when an azeotrope is boiled, the resulting vapor has the same ratio of constituents as the original mixture.”
Chemical Kinetics: Rates and Mechanisms of Chemical Reactions
Catalyst speeds up reaction but emerges unchanged
Speed and slow reactions
Conentrations of reactants
Reaction rate generally increases with higher concentration
Temperature
Reaction rates generally increas with temperature increase
Surface Area
Instead of solution and reactions in surfaces, rate increases as SA increase
Catalysis
Inhibitors slow down, catalysts speed up
Rate
Changes concentrations per change of time
Usually set rates as positive numbers
Rate of formation of produc or negative of the rate of disappearance of reactant.
General Rate of Reaction for the reaction
Obtained by dividing the rate of disappearance of reactorant or rate of formation of a product by soichiometric coefficient
aA + bB -> cC + dD
Rate = –1a[A]t = –1b[B]t=1c[C]t=1d[D]t
This is average rate because beinning of the interval, the instantaneous rate is faster than average.
Instantaneous rate of reaction is the limit as t goes to 0.
tangent line
Initial rate of reaction is instantaneous at beginning
Negative slope of tangent line for graph of reactants vs time to make reaction rate positive
Positive slope of tangent line for graph of products vs time
Rate Law of Chemical Reactions
For aA + bB -> cC + dD
Rate = –1a[A]t = –1b[B]t=1c[C]t=1d[D]t
Rate = k[A]m[B]n…
[A] and [B] are molearities of reactants at ceraain time
Exponents m and n are small positive inters (0,1,2,..), but may be negative or occasionally nonintegral
Exponents are determined by experiement
Value of exponents establish order of reaction
Overall order is sum of exponents
k is rate constant (proportionality constant)
Units for k
s-1 for first order
M s-1 for zero
M-1 s-1 for second
M-2 s-1 for thirde
General order : M(1 – overall order)s-1
Methods of Intial Rates
Find common two conentratiosn and find ratio of intial rates
Plug in and find ratio between the rates and get the exponent value
Effect on intial rate caused by doubling concentration of one reactant while concentration of other are held constant
Zeor order in reacant
Initial rate uncahnged
First order
Doubles
Second order
Quadruples
Third order
eight fold
First Order Reactions
Where single reactant yields product
Rate = k[A]1
Integrated rate law
Describes conecntration of a reactant as function of time
ln[A]t[A]0= -kt where t and 0 are subscripts
Used to find concentration of a reactant at a later time from inital conecntration
t is elpased time
Also as ln [A]t = -kt + ln [A]0
Variation for gases
P = constant * M
Half life
t1/2 of reaction is the time in which one half of reactant originally present is consumed.
For first order reaction A -> [A]t = 0.5*[A}0
A can be molarity, partial pressure, or mass
Integrated rate law
ln ( [A]t1/2 / [A]0 ) = -k*t1/2 = ln(½)
t1/2 = -ln(½) / k or 0.693/k
Half life is constant (depends on k and not concentration)
Other order Reactions
Zero order
Rate = k
Rates are the same for different concentrations
Rate is independent of concentration
[A]t = -kt + [A]0 integrated rate law
[A]t =0.5*[A]0 when reaction is half completed so t1/2 = [A]0/2k
Half life directly proportional to initial concentration
Second order
For single reactant: A -> product
Rate = k*[A]2
1/[A]t = kt + 1/[A]o
t1/2 = 1/(k*[A]0)
Half life depends on k and initial concentration

Summary of the Kinetics for Reactions of the Type aA → Products that are Zero, First, or Second Order in [A] | |||||||||||||||
Order | |||||||||||||||
Zero | First | Second | |||||||||||||
Rate law: | Rate = k | Rate = k[A] | Rate = k[A]2 | ||||||||||||
Integrated rate law: | [A] = –kt + [A]0 | ln[A] = –kt + ln[A]0 |
| ||||||||||||
Plot needed to give a straight line: | [A] versus t | ln[A] versus t |
| ||||||||||||
Relationship of rate constant to the slope of straight line: | Slope = –k | Slope = –k | Slope = k | ||||||||||||
Half-life: |
|
|
| ||||||||||||
Theories of chemical kinetics
Collision Theory
Reactions occurs from collision
Have to be fast molecules colliding
Activation energy (Ea) min energy to supplied from collision for a reaction
Collision frequency and fraction of activated species affect reaction rate
Most cases Orientation of colliding species also affects the rate
Ex. of orientation not matter is for H atom where every approach is same cause symmetrical distribution of electron cloud
Transition state theory
configuration of the atoms of colliding species at the crucial point of making products is the transition state
activated complex is the transitory species having this configuration.
transition state is like when quarterback and running back both holding the ball
activated complex has partial bonds between reactants and the parts that are broken up
reaction profile shows PE plotted in terms of progress of reaction
H = Ea(forward) – Ea(reverse)
Effect of Temp on Rxn Rates
Higher temps increase fraction of molecules with enough KE to produce reaction
percentage of effective collisions is greater increasing rates of rxn
For the rate constant k
k = Ae–Ea/RT
Svante Arrhenius equation
R is gas constant 8.3145 J mol-1 K-1
A is frequency factor (constant) and is product of collision frequency (Z) and a propability factor (p) accounting orientations
Zp = numbers of collisions per unit time
second part of equation is fration of molecular collisions suffient for rxn
ln k = –EaRT+ ln A
ln k is y, m = -Ea/R , 1/T = x , ln A = b for y = mx+b
Given rate constant at two temps
ln k2ln k1= EaR( 1T1– 1T2)
Reaction Mechanisms
reaction mechanisms is a series of simple steps that lead intital reacts to final products
elementary reaction represents each stage in progress of reaction meachnism
molecularity of an elementary rection is number of free atoms, ions entering the reaction
unimolecular is one molecule dissociate
bimolecular is two molecules colleid
termoleecular is three molecules colliding
exponents in the rate law is same as stoic coefficients in equation of elementary reaction
rate determining step is the slowest elementary reaction
the substance consumed in one step but not appearing in another step for the overall reaction is an intermediate
forward and revees rates are equal through rapid equilibrium
rate constant = k = (k1k2)/k-1 where k1 is rate constant for forward reaction, k-1 is the rate constant for reverse reaction and k2 is rate constant for original rate reaction with the spectator ion. The spectator ion is replaced with the net ionic portion. This is for fast reversible step followed by slow step
CHEMICAL EQUILIBRIUM
Dynamic nature of Equilibrium
In equilibrium, the forward reaction and reverse reaction proceed at equal rates and concentrations of reactants/products are constant
any reactions in which reactands and products attain constant nonzero conenctration is reversible
The Equilibrium Constatn Expression
Ratio of equilibrium concentrations raised to powers equal to the stoic coefficients is the equilibrium constant expression
Constant value denoted by Kc called concentration equilibrium constant
temperatures are noted
units are generally omitted
For equation a A + b B + …. g G + h H + …, equilibrium constant expression is Kc = [G]g[H]h….[A]a[B]b…. (products over reactants)
for 2 HI (g) H2(g) + I2(g) the kinetics kf[HI]2 =kr[H2][I2]. Therefore, kfkr=Kc
Keq is thermodynamic equilbruim constant
dimisionless quantities is activities
Modifying Equilibrium COnstant Expressions
modifying the chemical equation
RULES
When the equation for a chemical reaction with a equilibruium constant Kc is reversed, the equilibrium constant is inverted. The reverse reaction has equilibrium constat of K’c = 1/Kc
When coefficients of an equation are multiplied by a common factor n to produce a new equation, we raise the original Kc value to the power n to obtain the new equilibrium constant.
A balanced chemical equation should be written when citing a value for Kc because the constant depends on how the equation is written
equilibrium constant for overall reaction
When we add the equations for individual reactions to obtain an overall equation, we mutliply theire equilibrium constants to obtain the equilibrium constant for overall reaction.
Equilibria involving gases
Easier to measure partial pressure
For equation a A + b B + …. g G + h H + …, partial pressure equilibrium constant expression is Kp = [PG]g[PH]h….[PA]a[PB]b…. (products over reactants)
Given Kc, Kp = Kc*(RT)ngaswhere ngas = (g+h+…) – (a+b+…) which is the products minus reactants coefficients
Equilibria involving pure solids and liquids
Heterogeneous reactions: reactants and products not in same phase
Homogeneous reactions: all reactants/products are gases
Equilibrium constant expressions do not include terms for any reactants or products present as pure solids or pure liquids because their concentrations do not change in a reaction
Equilibrium constants: when to use?
A very large numerical value of Kc or Kp signifies reaction goes to completion (In effect, not reverisble)
A very small numerical value of Kc or Kp signifies that the forward reaction, as written, occurs only to a very slight extent or does not occur.
Usually described as no reaction
Thermodynamically favorable is when equilibrium constant is large but is kinetically controlled ( meaning that the exceedingly slow reaction rate prevents significant reaction for occuring)
Such as hydrogen and oxygen gas to make water at room temp
An equilibrium constant expression does not fully describe the extent of a chemical reaction under all conditions at a given time. In some cases, the reaction is limited by chemical kinetics.
If K is about equal to 1, the reaction will reach equilibrium as an intermediate mixture, meaning the amounts of products and reactants will be about the same.
Reaction quotioent Q: direction of net change
For nonequilibrium conditions, the reaction quotient (Qc or Qp) is the expression having the same form as the concentrations ratio or partial pressures ration from Kp or Kc (basically same eqaution but for concentrations not a equilibrium)
Not constant
If Qc < Kc , a net change occurs in the forward direction which is from left to right in the written equation for the reaction. ( The rate of the forward reaction exceeds that of the reverse reaction until equilibrium is reached)
If Qc > Kc a net change proceeds in the reverse direction which is from right to left in the written equation for the reaction. (The rate of the reverse reaction exceeds that of the forward reaction until equilibrium is reached).
Henri Le Chatelier’s Principle
When any change in concentration, temperature, pressure, or volume is imposed on a system at equilibrium, the system responds by attaining a new equilibrium condition minimizing the impact of the changes
Concentrations
When the concentration of a product is increased, the reaction proceeds in reverse to decrease the concentration of the products.
When the concentration of a reactant is increased, the reaction proceeds forward to decrease the concentration of reactants.
If pure solid or liquid is added, no change in equilibrium condition
Pressures or Volume
increase in pressure or a decrease in volume causes the reaction to shift to the side with less gas moles
decrease in pressure or an increase in volume causes the reaction to shift to the side with more gas moles
no difference in number of moles of gas in a reaction then changes in pressure or volume have no effect on equilibrium
at constant pressure and inert gas added and volume expands to accommodate added gas, same effect as increasing volume
constant volume and adding a gas, equilibrium is not affected
Change Temp
Only change that changes equilibrium constant
if a temperature change causes the eq constant to increase, the reaction shifts right. if eq constant decrease, the reaction shifts left
Raising the temperature of an equilibrium mixture shifts equilibrium in the direction of an endothermic reaction to absorb the heat
Lowering the temperature shifts equilibrium in the direction of the exothermic reaction to gain some of the heat lost.